Amyotrofisk lateral sklerose - Årsaker, symptomer og behandling

Amyotrofisk lateralsklerose (ALS), også kjent som motornervesykdom eller Lou Gehrigs sykdom, er en alvorlig nevrodegenerativ sykdom (en sykdom som forårsaker degenerasjon av nevroner) av ukjent årsak og uten kurativ behandling.
Amyotrofisk lateralsklerose er en sykdom som forårsaker progressivt tap av styrke og atrofi av muskler, noe som fører til døden på rundt 3 til 5 år. Det er imidlertid tilfeller med lengre overlevelsestid. 10% av pasientene med ALS kan leve i mer enn 10 år, og en svært liten andel kan leve i mer enn 20 år hvis sykdommen sparer musklene som er ansvarlige for å puste og pasienten kan motta intensiv behandling.
Det mest kjente tilfellet av lang levetid med amyotrofisk lateralsklerose er den engelske fysikeren Stephen Hawking, som hadde sykdommen diagnostisert 21 år og lever fortsatt og produktiv over 70 år. Dessverre er saker som hans svært sjeldne og representerer ikke den vanlige utviklingen av ALS.
I denne artikkelen vil vi forklare hva som er amyotrofisk lateralsklerose, hva er årsakene og risikofaktorene, dens symptomer, hvordan diagnosen er gjort og hva er alternativene til behandlinger som eksisterer hittil.
Hva er motor neuron
For å forstå hva amyotrofisk lateral sklerose er, som, som allerede nevnt, også kalles motor neuron sykdom, er det først nødvendig å vite hvordan samspillet mellom nerver og muskler fungerer. La oss forklare dette forholdet på en veldig kort og didaktisk måte.
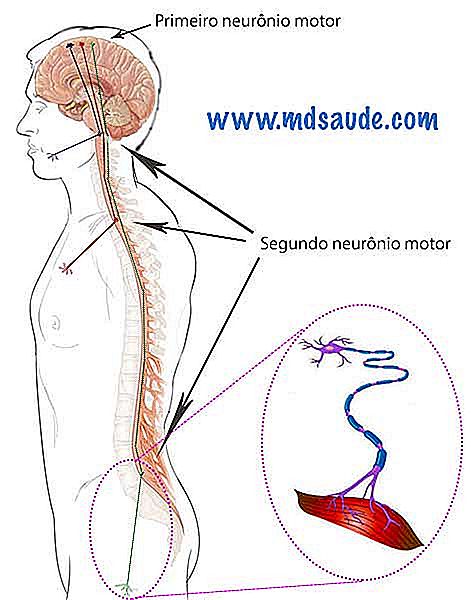
Eventuell frivillig muskelkontraksjon utføres takket være aktiveringen av en gruppe neuroner som kalles "motorneuron". Hvis du beveger fjærene dine, løfter armene dine, senker hodet eller bestemmer deg for å sitte, alle disse bevegelsene ble født inne i hjernen din, som ved hjelp av elektrisk stimulering gjorde meldingen til de muskelklyngene du har tenkt å bruke.
Det er 2 typer motorneuron: øvre motorneuron, også kalt den første motorneuron, og den lavere motorneuron, kjent som den andre motorneuron.
Den første motorneuron ligger i hjernen, i et område kalt cerebral cortex. For enhver frivillig kontraktsmusikk å bevege seg, gir den første neuronen en elektrisk impuls som beveger seg gjennom hjernen, passerer gjennom hjernestammen og når ryggmargen som befinner seg i ryggraden. I ryggmargen er det andre motorneuronet, som stimuleres ved å motta den elektriske impulsen fra den første nevronen. Den andre motorneuronen gir opphav til perifere nerver som fordeles av kroppen for å innervative musklene.
Ryggmargen opptar hele den sentrale kanalen i ryggraden, går fra livmorhalsen (nakke) til begynnelsen av lumbale ryggraden. Kroppens områder er innervaret fra topp til bunn, det vil si at den andre motorneuronene som innerverer de øvre lemmer, befinner seg i den cervicale ryggraden, mens den andre motorneuronene som innerverer underbenene ligger under, i høyden av den første lumbale vertebrae.
Merk: I tilfelle av kranialnervene, som innerverer ansiktsmuskler, som øyne og munn, er den andre motorneuronen i hjernestammen, en region like under hjernen.
Når en person lider av en ulykke med alvorlig skade på ryggmargen, mister den nøyaktig den kontakten mellom den første motorneuron i hjernen og den andre motorneuron i ryggmargen. Hvis ryggmargen er på høyden av livmorhalsen, mister pasienten bevegelsene i nakken ned. Hvis lesjonen er lavere på slutten av thoracic ryggraden eller lumbale ryggraden, mister den bare bevegelsene på underdelene.
Hva er amyotrofisk lateral sklerose?
Amyotrofisk lateralsklerose er en sjelden sykdom som påvirker omtrent 0, 002% av befolkningen. I Brasil er det ca 6000 mennesker med denne sykdommen.
ALS oppstår på grunn av degenerasjon av det første motorneuron og / eller det andre motorneuron. Pasienten presenterer progressivt tap av styrke i alle muskelgrupper hvis innerverende nevroner lider av degenerasjon.
ALS påvirker bare musklene med frivillig sammentrekning, det vil si de musklene som bare beveger seg når vi vil. Muskler i hjertet og tarmene, for eksempel, er ufrivillig sammentrekning og er dermed spart i amyotrofisk lateralsklerose. Åndedrett, til tross for at det utføres ufrivillig, blir mesteparten av tiden støttet av muskler av frivillig sammentrekning, og derfor kan vi holde pusten i noen sekunder. Derfor er lammelse av respiratoriske muskler et vanlig tegn i ALS, som er den faktoren som vanligvis bestemmer pasientens død.
Begrepet "amyotrofisk" betyr muskelatrofi, noe som er en av konsekvensene av mangel på sammentrekning på grunn av fravær av nervestimulering. De muskelregioner som er berørt av sykdommen lider atrofi. Begrepet sklerose kommer fra det faktum at degenerasjon av nevroner fører til ødeleggelse, disse nervecellene blir erstattet av en slags arrvæv. For å fullføre, kalles sykdommen amyotrofisk lateral sklerose fordi den andre motorneuronen vanligvis forblir i den mer laterale regionen av margen, som lider av sklerose med utviklingen av sykdommen.
Selv om det ofte kalles motor neuron sykdom, er sannheten at amyotrofisk lateral sklerose er bare en av 4 eksisterende former for denne typen nevrologisk sykdom. ALS er den vanligste formen, og står for om lag 70% av tilfellene med motornervesykdom. I tillegg til ALS er det også følgende sykdommer:
- Progressiv muskelatrofi - som er skjemaet som bare påvirker det andre motorneuronet og tilsvarer ca 10% av tilfellene.
- Progressiv bulbar lammelse - tilsvarer 20% av tilfellene og er vanligvis begrenset til kranmotoriske nerver. Denne formen påvirker den første og andre motornerven.
- Primær lateral sklerose - er den mest sjeldne, men godartede formen for motorneuronsykdom. Det er vanligvis begrenset til det første motorneuronet og har svært langsom progresjon.
De 4 formene av motornervesykdom bør ikke betraktes som forskjellige sykdommer, men heller variasjoner i presentasjonen av en enkelt sykdom. Pasienten kan for eksempel i utgangspunktet ha et mer suggestivt bilde av progressiv bulbarlammelse og senere utvikle et typisk ALS-bilde.
Risikofaktorer for amyotrofisk lateral sklerose
Vi vet ikke hvorfor sykdommen ser ut i mer enn 90% av tilfellene. På bare 5 til 10% er det et klart genetisk forhold, og ALS overføres på kjent måte.
Selv om de eksakte årsakene er uklare, er det allerede kjent noen risikofaktorer. ALS er en sykdom som rammer personer over 40 år, med en toppsituasjon rundt 75 år. Det er imidlertid tilfeller fra begynnelsen av ungdomsårene. Menn er 2 ganger mer berørt enn kvinner.
Røyking er en kjent risikofaktor. I tillegg til å doble risikoen for å utvikle amyotrofisk lateral sklerose, har røykere en tendens til å ha en mer aggressiv form for sykdom. Den gode nyheten er at pasienter som er i stand til å slutte, har samme risikofaktor som folk som aldri har røykt.
Kontinuerlig eksponering for visse stoffer, spesielt tungmetaller, som bly, synes også å være en risikofaktor.
Av grunner som ikke er godt forstått, har profesjonelle idrettsutøvere, for eksempel fotballspillere, en høyere frekvens av ALS i forhold til den generelle befolkningen. Det antas at intens muskelopplæring kan være en utløser for skade på motornerven i genetisk forutsettede mennesker.
Som i utøvere er amyotrofisk lateral sklerose også mer vanlig hos militære menn. Årsakene er ennå ikke klargjort.
Symptomer på amyotrofisk lateral sklerose
Symptomene på ALS forekommer ved progressiv ødeleggelse av motorneuroner, enten de er overlegen eller dårligere. De vanligste innledende symptomene er progressiv svakhet i hendene, vanskeligheter med å gå, mangel på styrke i føttene, vanskeligheter med å snakke eller svelge, kramper eller myofascikulasjoner (små, raske og ufrivillige bevegelser av muskler som kan bli lagt merke til gjennom hud eller tunge ).
ALS forårsaker ikke endringer i funksjonen av hjertemuskulatur, blodårer, blære eller mage-tarmkanalen. I senere stadier kan pasienten imidlertid bli forstoppet, ikke på grunn av svakhet i tarmmuskulaturen, men på grunn av mangel på styrke i bukmuskulaturen, noe som bidrar til utvisning av avføringen. Takt, hørsel, smak og lukt blir også ofte bevart, spesielt i de tidlige stadiene av sykdommen. Seksuell funksjon er vanligvis ikke påvirket, men etter hvert som sykdommen utvikler seg, på grunn av alle komplikasjoner, opphører pasientens seksuelle interesse å eksistere.
Den vanligste formen for å åpne rammen er asymmetrisk svakhet, som bare påvirker en hånd eller ben. Svakhet i hender blir ofte notert fordi pasienten har problemer med å holde gjenstander, for eksempel et glass. Det er også vanlig å utføre oppgaver som krever finere koordinering, for eksempel å trykke på en knapp eller sette nøkkelen i nøkkelhullet. Svakhet i underkroppene manifesteres vanligvis av vanskeligheten eller følelsen av at det er noe rart å gå eller løpe. I begynnelsen kan symptomene på amyotrofisk lateralsklerose være subtil, noe som gjør at pasienten forsinker å søke medisinsk hjelp.
Med sykdomsprogresjonen har symptomer som har startet opp på en lokal måte en tendens til å bli spredt av flere muskler i kroppen. Pasienten presenterer symptomer på skade på første og andre motorneuroner, som kan identifiseres ved fysisk undersøkelse. For eksempel er muskelstivhet, tap av motorisk koordinasjon og økte reflekser (identifisert av den berømte knehammertesten) tegn på første motorisk nevronesykdom. Imidlertid er svakhet, muskelatrofi, tap av reflekser, myofaskikulasjoner og kramper symptomer på skade på den andre motorneuron.
Forsiktig, vær ikke forsiktig, fordi kramper (les: Kramper | Årsaker og behandling) og myofaskikulasjoner er vanlige funn som kan forekomme hos noen. Myofaskikulasjoner av tungen er imidlertid typiske for ALS.
I de fleste tilfeller, til tross for vanskeligheten ved å snakke, forblir den intellektuelle delen av pasienten praktisk talt intakt. Han har full oppfatning av hva som skjer og forstår perfekt folkene rundt seg. I omtrent 15% av tilfellene kan imidlertid demens utvikle seg sammen med motorens symptomer på ALS.
I de avanserte stadier av sykdommen blir pasienten fullstendig lammet. Innånding av pustemuskulatur kan forekomme tidlig, men det er vanligvis et tegn på avansert sykdom. Åndedretts komplikasjoner ende opp med å være hovedårsaken til død av pasienten med amyotrofisk lateral sklerose.
Diagnose av amyotrofisk lateral sklerose
Ingen enkel test eller undersøkelse er i stand til å etablere den endelige diagnosen av amyotrofisk lateral sklerose. Diagnosen av ALS er hovedsakelig basert på symptomer og tegn som er observert gjennom fysisk undersøkelse. Tilstedeværelsen av både første og andre motorneuronsignaler på samme tid er ganske suggestiv.
Siden symptomene på ALS i de tidlige stadier kan lignes på flere andre sykdommer, krever nevrologen ofte komplementære tester for å utelukke differensialdiagnoser. En av disse testene er elektro-urografi, en undersøkelse som kan avgjøre om kilden til muskel svakhet er muskel selv eller noen sykdom i nerver. Legen kan også bestille en MR for å fange detaljert bilder av hjernen og ryggmargen for å utelukke andre årsaker til nevrologiske sykdommer som svulster, slag, infeksjoner, hernierte plater etc.
Behandling av amyotrofisk lateral sklerose
Dessverre er det fortsatt ingen kur for amyotrofisk lateral sklerose. Behandlingen tar derfor sikte på å redusere sykdomsprogresjonen, forhindre komplikasjoner og forbedre livskvaliteten. Fysioterapi, psykologisk støtte, taleterapi og ernæring er avgjørende for å nå disse målene.
Hos pasienter med åndedrettsstød kan bærbare ventilatorer, kalt ikke-invasiv positivtrykksventilasjon, brukes til å hjelpe pasientene å puste mer effektivt. I begynnelsen kan enheten brukes under søvnen, og som sykdommen utvikler seg gjennom dagen.
Hos pasienter som har problemer med å mate på grunn av manglende evne til å svelge mat, er perkutan gastrostomi et av alternativene. Denne behandlingen består i plassering av et rør gjennom huden direkte til magen, og blir denne ruten for matadministrasjon.
Pasientens overlevelsestid avhenger veldig mye av pasientens tilgang til hele medisinsk apparat, inkludert hjelp fra mange helsepersonell.
Rettsmidler for amyotrofisk lateral sklerose
Det er bare en godkjent medisin på markedet for behandling av ALS. Riluzole, solgt under handelsnavnet Rilutek, er det eneste stoffet som har vist seg å øke levetiden til pasienter med amyotrofisk lateralsklerose. Det er imidlertid ikke et mirakelmedisin, overlevelsesøkningen er liten, omtrent 6 måneder til 1 år i gjennomsnitt.

VIDEO: 10 årsaker til tidlig evakuering
Besøk vår Youtube-kanal: https://www.youtube.com/mdsaude Video transkripsjon Hei, har du det travelt? Så det bremser for oss å snakke om for tidlig utløsning. Visste du at nesten alle årsaker er psykologiske? I denne videoen vil jeg presentere ti grunner som fører at en mann skal ekkulere fort. 1- BE

FLUCONAZOL - Behandling for candidiasis og mykoser
Flukonazol er et antifungalt stoff av den triazoliske familien, mye brukt i behandling av forskjellige mykoser, spesielt de som er forårsaket av Candida-sopp, slik som vaginal candidiasis eller candidiasis av oropharynx, populært kjent som trussel. Fluconazol kan gis oralt eller intravenøst. Sistnevnte skjema er vanligvis reservert for alvorlige tilfeller. F



